201904
News > Newsletter Archiv
Inhalt Newsletter April 2019
Computerspiele können bei Kleinhirnpatienten Koordination verbessern
© hil/aerzteblatt.de
https://www.aerzteblatt.de/fachgebiete/neurologie/news?nid=97430
Tübingen – Auf die Möglichkeiten von Health Games hat das Hertie-Institut für klinische Hirnforschung (HIH) in Tübingen hingewiesen. „Unsere Studien haben ergeben, dass Patienten, die durch Schäden am Kleinhirn unter Bewegungsstörungen leiden, durch das Training mit speziellen Videospielen ihre Beweglichkeit verbessern können“, erläuterte Matthis Synofzik aus dem HIH anlässlich der Computerspielemesse Gamescom in Köln.
Die Arbeitsgruppe um Synofzik und den Bewegungswissenschaftler Winfried Ilg entwickelt an dem Institut spezielle Videospiele für Menschen mit einer degenerativen Ataxie, die durch genetisch bedingte Schäden am Kleinhirn entsteht. Die Betroffenen leiden vor allem unter Bewegungsstörungen wie ungelenken Armbewegungen, Gangunsicherheit und häufigen Stürzen.
Regelmäßige Krankengymnastik kann eine Verschlechterung hinauszögern. Doch vor allem Kinder und junge Erwachsene sind laut Synofzik oft nicht ausreichend für eine Physiotherapie zu begeistern und trainierten nicht so häufig, wie sie sollten. Zudem reiche die von der Krankenkasse verschriebene Physiotherapie für diese Patienten nicht aus und werde auch Zuhause oft nicht fortgeführt. „Um das Motivationsproblem zu lösen kamen wir auf die Idee, Videospiele zu verwenden, die mit ganzem Körpereinsatz gesteuert werden und die man zum Training verwenden kann, sogenannte Exergames“, erläuterte Synofzik.
„Wir haben mit einem Tischtennisspiel begonnen, dort waren die Anforderungen auf einfachster Stufe so gering, dass auch Patienten mit Koordinationsstörungen Erfolgserlebnisse hatten“, berichtet Ilg, Leiter des Klinischen Bewegungslabors der Abteilung Kognitive Neurologie am HIH. Weitere Spiele, in denen die Kinder und jungen Erwachsenen in anspruchsvoller Körperhaltung virtuelle Wassertanks abdecken oder mit dynamischem Ausfallschritt auf Leuchtflächen reagieren müssen, kamen hinzu.
Selbst bei schwer betroffenen Patienten konnten die Wissenschaftler die Symptomatik verbessern. „Eine Ataxie wird bei einem Erkrankten auf einer Skala von 0 bis 40 im Mittel um 1,2 Punkte pro Jahr schlechter. Durch vier bis sechs Wochen Training haben unsere Patienten zwei Punkte gewonnen. Das heißt salopp gesagt: Sie haben ungefähr eineinhalb Jahre Krankheit wettgemacht“, berichtet Synofzik.
Die Arbeitsgruppe weist darauf hin, dass das Kleinhirn aufgrund vieler Erkrankungen geschädigt sein könne, zum Beispiel durch einen Tumor, Schlaganfall, Multiple Sklerose oder durch altersbedingten Abbau. „Da wird der Markt auf einmal riesig. Vielleicht erreichen diese Videospiele irgendwann sogar die Marktreife als zugelassenes Medizinprodukt. Das wäre dann Gaming auf Rezept“, so Synofzik.
Neuroprotektion nach Schädel-Hirn-Trauma: Frühzeitige Hypothermie ist klinisch ohne Nutzen
Autor: Gießelmann, Kathrin
Dtsch Arztebl 2018; 115(46): A-2128 / B-1762 / C-1740
Cooper D J, Nichol A D, Bailey M, et al.: Effect of Early Sustained Prophylactic Hypothermia on Neurologic Outcomes Among Patients With Severe Traumatic Brain InjuryThe POLAR Randomized Clinical Trial. JAMA 2018; doi: 10.1001/jama.2018.17075
aus: https://www.aerzteblatt.de/archiv/202798/Neuroprotektion-nach-Schaedel-Hirn-Trauma-Fruehzeitige-Hypothermie-ist-klinisch-ohne-Nutzen
Erneut kommt eine multizentrische Studie zu dem Ergebnis, dass die Hypothermie nach traumatischer Hirnverletzung keinen Nutzen hat. Für die Autoren um Jamie Cooper und Alistair Nichol von der Monash University in Melbourne ist die Strategie damit gescheitert.
Etwa 50–60 Millionen Menschen weltweit erleiden jedes Jahr eine traumatische Hirnverletzung (TBI) und mehr als die Hälfte der Weltbevölkerung wird mindestens ein TBI im Laufe ihres Lebens erleiden. Schon lange diskutieren Experten kontrovers über die Vorteile der Kühlung des Gehirns in der Intensivstation nach einer TBI. Die Theorie: Die Abkühlung oder Unterkühlung reduziert die Enzephalitis und daraus folgende Hirnschäden.
Die POLAR-Studie mit 466 Patienten kann diese Theorie nicht bestätigen. Sie kommt zu dem Schluss, dass auch die frühzeitige und prophylaktische Hypothermie das klinisch-neurologische Endergebnis von Patienten mit schwerem Schädel-Hirn-Trauma nicht verbessert.
Mit der Hypothermie begannen die Ärzte durchschnittlich 1,8 Stunden nach der Verletzung, die Wiedererwärmung erfolgte langsam nach etwa 22,5 Stunden. Günstige Ergebnisse gemessen am Glasgow Outcome Scale traten nach 6 Monaten bei 117 Patienten (48,8 %) in der Hypothermie-Gruppe und bei 111 (49,1 %) in der Kontrollgruppe auf. Das relative Risiko unterschied sich in beiden Gruppe nicht signifikant (RR = 0,99 [95 % CI, 0,82– 1,19]; p = 0,94).
Pneumonien und intrakranielle Blutung traten in den ersten 10 Tagen nach der Hypothermie etwas häufiger auf: Raten der Pneumonie 55,0 % gegenüber 51,3 %, und die Raten der erhöhten intrakraniellen Blutung betrugen 18,1 % gegenüber 15,4 %.
Fazit: „Dies ist die 4. große randomisierte multizentrische Studie zum Thema ‚Hypothermie nach schwerem Schädel-Hirn-Trauma‘, die ein negatives Ergebnis zeigt“, resumiert Prof. Dr. med. Andreas Unterberg, Direktor der Klinik und Poliklinik für Neurochirurgie am Universitätsklinikum Heidelberg. Das sei umso bedauerlicher, als zuvor viele experimentelle Studien in unterschiedlichsten Modellen eindeutige neuroprotektive Wirkungen der Hypothermie erbracht hatten. „Die derzeit einzig wirksame, evidenzbasierte, neuroprotektive Methode beim schweren Schädel-Hirn-Trauma ist die dekompressive Kraniektomie bei therapieresistenter intrakranieller Drucksteigerung.“
Morbus Parkinson: Früher Behandlungsbeginn mit L-Dopa in Studie ohne Einfluss auf Krankheitsverlauf
aus https://www.aerzteblatt.de/fachgebiete/neurologie/studien?nid=100630
© rme/aerzteblatt.de
Amsterdam – Die Behandlung des Morbus Parkinson mit L-Dopa, seit 4 Jahrzehnten das Standardmedikament der Erkrankung, ist rein symptomatisch. In einer randomisierten Doppelblindstudie aus den Niederlanden, die jetzt im New England Journal of Medicine (2019; 380: 315-324) publiziert wurde, konnte weder eine günstige krankheitsmodifizierende Wirkung nachgewiesen werden, noch bestätigte sich die verbreitete Befürchtung einer toxischen Wirkung des Wirkstoffs.
L-Dopa ist zu Beginn der Erkrankung ein sehr wirksames Medikament. Die motorischen Symptome der Patienten verbessern sich rasch. Nach einiger Zeit kommt es jedoch erneut zu einer Verschlechterung und die Behandlung wird durch das Auftreten von Fluktuationen und Dyskinesien kompliziert. Dies hat zu der Vermutung geführt, dass L-Dopa eine toxische Wirkung hat. Viele Neurologen raten den Patienten deshalb, nicht sofort mit einer Therapie zu beginnen. Wer in der ersten Zeit die Symptome erträgt, hat nach dieser Annahme später einen längeren Nutzen von der Therapie.
Auf der anderen Seite gibt es die Vermutung, dass L-Dopa nicht nur den fehlenden Neurotransmitter ersetzt, sondern darüber hinaus das Fortschreiten der Erkrankung verlangsamt. Dafür hatte vor Jahren die ELLDOPA-Studie Argumente geliefert (NEJM 2004; 351: 2498-2508). In der randomisierten Studie waren Patienten über 40 Wochen mit L-Dopa (in unterschiedlicher Dosis) oder Placebo behandelt worden. Danach waren in beiden Gruppen die Medikamente abgesetzt worden. In den L-Dopa-Gruppen kam es zwar zu einer Verschlechterung. Die Symptome erreichten in den ersten Wochen jedoch nicht den Schweregrad der Placebogruppe. War dies eine Nachwirkung von L-Dopa oder hatte das Medikament tatsächlich den Verlauf der Erkrankung günstig beeinflusst?
Der „Delayed-start Trial Levodopa in Early Parkinson’s Disease“ (LEAP) sollte diese Frage klären. An 57 Kliniken in den Niederlanden wurden 445 Patienten mit neu diagnostiziertem Morbus Parkinson in der ersten Phase über 40 Wochen auf eine Behandlung mit L-Dopa/Carbidopa (100mg/25 mg 3-mal täglich) einschließlich einer Dosissteigerung von 2 Wochen, oder auf eine 40-wöchige Placebotherapie (3-mal täglich) randomisiert. In der zweiten Phase in den Wochen 40 bis 80 erhielten dann alle Patienten L-Dopa/Carbidopa (100mg/25 mg 3-mal täglich).
Primärer Endpunkt war der Unterschied in der Gesamtpunktzahl der „Unified Parkinson Disease Rating Scale“ (UPDRS) am Studienende nach 80 Wochen. Die UPDRS bewerte die Symptome von 0 bis 176 Punkten, wobei eine höhere Punktzahl eine schwerere Krankheit anzeigt.
Zu Studienbeginn hatten die 222 Patienten der Frühstartergruppe einen UPDRS von im Mittel 28,1 Punkten gegenüber 29,3 Punkten in der Spätstartergruppe. Wie das Team um Rob de Bie vom Medizinischen Zentrum der Universität Amsterdam berichtet, kam es in der Frühstartergruppe in den ersten 40 Wochen zu einer Verbesserung des UPDRS um 3,1 Punkte gegenüber einer Verschlechterung um 2,0 Punkte in der Placebogruppe. Die Differenz von 5,1 Punkten war mit einem 95-%-Konfidenzintervall von 2,9 bis 7,2 Punkten signifikant.
In der zweiten Phase der Studie holten die Spätstarter jedoch auf. Nach 80 Wochen hatten sie gegenüber dem Ausgangswert 2,0 Punkte verloren gegenüber einem Verlust von 1,0 Punkten in der Frühstartergruppe. Die Differenz von 1,0 Punkten war mit einem 95-%-Konfidenzintervall von minus 1,5 bis 3,5 Punkte nicht signifikant.
Damit haben sich die Hoffnungen auf eine positive krankheitsmodifizierende Wirkung von L-Dopa zerschlagen. Aber auch die Befürchtung, dass ein früher Behandlungsbeginn den Verlauf der Erkrankung beschleunigt, hat sich nicht bestätigt.
Die klinische Bedeutung der Ergebnisse ist enorm, findet der Parkinsonexperte und Mitautor Günther Deuschl vom Universitätsklinikum Schleswig-Holstein in Kiel. Die Toxizitätsdiskussion um L-Dopa sei damit so gut wie beendet, schreibt Deuschl in einer Pressemitteilung der Deutschen Gesellschaft für Neurologie.
Parkinson: Warum eine lange L-Dopa-Therapie Dyskinesien verursacht
Einen weiteren Einwand könne die Studie allerdings nicht widerlegen. Es sei durchaus möglich, dass bei einem früheren Therapiebeginn die Fluktuationen und Dyskinesien früher auftreten. Dem stehe allerdings ein deutlicher Gewinn an Lebensqualität in den ersten Jahren der Erkrankung gegenüber, der mit L-Dopa erreicht werde.
Nach Einschätzung der Deutschen Gesellschaft für Neurologie hat die LEAP-Studie Zweifel ausgeräumt und gezeigt, dass die bewährte L-Dopa-Therapie langfristig sicher ist. Dennoch sei die Suche nach weiteren, neuen Therapien beim Morbus Parkinson besonders dringlich. Nach einer Analyse der „Global Burden of Disease“- Studie hat sich die Zahl der an Parkinson erkrankten Menschen seit 1990 mehr als verdoppelt, was überwiegend auf die zunehmende Lebenserwartung der Bevölkerung zurückzuführen ist.
Erstes Medikament zur Behandlung der Muskeldystrophie Duchenne zugelassen
von Treat-NMD
aus https://www.treat-nmd.de/register/clinical_trials/dmd/ataluren_zulassung/index.de.html
Wir möchten Sie heute über das Patientenregister darüber informieren, dass das Medikament Translarna™ (Ataluren) der Firma PTC Therapeutics zur Behandlung von Duchenne Muskeldystrophie-Patienten im Alter ab fünf Jahren mit Nonsense-Mutation von der Europäischen Union die bedingte Marktzulassung erhalten hat.
Es handelt sich dabei um ein Medikament, das spezielle Mutationen im Dystrophin-Gen behandelt, die zu einem Stopp der Ablesung des Gens und der Proteinproduktion in der Muskelzelle führen. Somit wird bei den betroffenen Duchenne-Jungen kein Dystrophin-Eiweiß in der Muskelzelle gebildet und es kommt zur Muskelschwäche. Diese Mutationen heißen Nonsense-Mutationen.
Die meisten Mutationen, die bei Duchenne-Jungen vorliegen sind sog. Deletionen oder Duplikationen, bei denen genetische Informationen doppelt oder gar nicht vorliegen (ausgeschnitten wurden). Hier liegt ein anderer genetischer Fehler zugrunde als bei den Nonsense-Mutationen, die zum Stopp führen; Patienten mit Deletionen und Duplikationen sprechen nicht auf die Therapie mit Translarna™ an.
Bei ca. 13% aller Duchenne-Patienten liegt eine Nonsense-Mutation vor und genau diese Patienten können zukünftig mit Translarna™ behandelt werden. Das Medikament versucht, den mutierten Bereich in der genetischen Information wie ein Pflaster zu bedecken, so dass dieser Fehler „überlesen“ wird und am Ende das Dystrophin-Eiweiß in der Muskelzelle (zu einem geringeren Anteil) gebildet werden kann.
Die Zulassung des Medikaments beruht auf einer weltweiten Studie mit 174 gehfähigen Patienten im Alter von über fünf Jahren, die über 48 Wochen durchgeführt wurde. Bei Patienten, die dreimal täglich mit einer Gesamtdosierung von 40 mg/kg/Tag behandelt wurden, nahm die durchschnittliche Gehstrecke im 6-Minuten-Gehtest nach 48 Wochen nur um 12,8 Meter ab – im Vergleich zu einer durchschnittlichen Verschlechterung der Gehstrecke um 44,1 Meter bei den Patienten mit Placeboeinnahme (Scheinmedikament). Mit diesem besseren Erhalt der Gehstrecke bei den behandelten Patienten konnte eine Verlangsamung des Erkrankungsverlaufs nachgewiesen werden, so dass die Zulassung genehmigt wurde.
Aufgrund der medizinischen Dringlichkeit und den vorliegenden Studiendaten haben die Europäischen Behörden eine sogenannte bedingte Zulassung für Translarna™ ausgesprochen – das heißt, dass PTC Therapeutics sich verpflichtet die vorliegenden Ergebnisse zur Wirksamkeit und Sicherheit in einer zweiten großen Studie zu bestätigen. Derzeit findet eine weitere 48-wöchige Studie mit 220 Patienten in 54 Zentren weltweit statt (ACT DMD), die erste Ergebnisse im Herbst 2015 liefern soll.
Die Ergebnisse zu Nebenwirkungen zeigten, dass das Medikament im Allgemeinen gut vertragen wurde. Ernsthafte Nebenwirkungen waren selten und waren nicht mit der Einnahme einhergehend. Die häufigsten Nebenwirkungen bei der empfohlenen Dosis waren Übelkeit, Erbrechen und Kopfschmerzen.
Zugelassen ist das Arzneimittel für Betroffene, die gehfähig und mindestens fünf Jahre alt sind. Die Nonsense-Mutation sollte mittels genetischer Testung bestätigt sein. Nur Spezialisten, die in der Behandlung von Duchenne-Patienten erfahren sind, sollten die Therapie verordnen. Das Medikament wird voraussichtlich Ende 2014/Anfang 2015 auf dem deutschen Markt erhältlich sein wird.
Alle im Patientenregister eingetragenen Patienten haben von uns heute diese Information per E-Mail/Post erhalten und sie wurden auch informiert, ob sie laut der Befunde im Patientenregister die für die Behandlung mit Translarna™ passende Mutation haben. Sollten Sie nicht sicher sein, ob eine Behandlung für Sie in Frage kommen kann, besprechen Sie dies bitte mit Ihrem Arzt. Eine genetische Untersuchung des Dystrophin-Gens ist zwingend nötig, damit über die Behandlungsmöglichkeit entschieden werden kann.
Die Erforschung von Medikamenten für Patienten mit anderen Mutationen geht ebenfalls weiter und klinische Studien werden durchgeführt. Wir danken auch Ihnen durch Ihre Teilnahme am Patientenregister, dies möglich zu machen. Über das Patientenregister konnten Patienten gefunden werden, die an früheren und derzeitigen Studien teilgenommen haben/teilnehmen. Nur dadurch kann die Wirksamkeit eines Medikaments gezeigt werden und zukünftig hoffentlich zu weiteren Zulassungen führen!
Bei Fragen wenden Sie sich bitte an Ihren behandelnden Arzt, da er in Ihrem speziellen Fall das Wissen hat, um eventuelle Entscheidungen zu treffen.
Polyneuropathien
Ursachen, Diagnostik und Therapieoptionen
Polyneuropathies—etiology, diagnosis, and treatment options
Sommer C, Geber C, Young P, Forst R, Birklein F, Schoser B: Polyneuropathies—etiology, diagnosis, and treatment options. Dtsch Arztebl Int 2018; 115: 83–90. DOI: 10.3238/arztebl.2018.0083
Hintergrund: Polyneuropathien sind in Abhängigkeit vom Alter mit einer Prävalenz von circa 5–8 % in der erwachsenen beziehungsweise älteren Bevölkerung die häufigsten Erkrankungen des peripheren Nervensystems. Therapeutische Optionen hängen entscheidend von den spezifischen Ursachen ab. Diese sollen daher durch die Diagnostik möglichst genau identifiziert werden.
Methode: Die Arbeit basiert auf den aktuellen Leitlinien und einer selektiven Literaturrecherche in PubMed nach großen Kohortenstudien sowie randomisierten kontrollierten Studien von 2000–2017 mit Fokus auf den nichthereditären Formen der Polyneuropathie.
Ergebnisse: Diabetes mellitus ist die häufigste Ursache von Polyneuropathien in Europa und Nordamerika. Die Alkohol-assoziierte Polyneuropathie hat eine Prävalenz von 22–66 % unter chronisch Alkoholkranken. Aufgrund der Zunahme maligner Erkrankungen und neuer Substanzen in der Tumorbehandlung sind auch Chemotherapie-induzierte Neuropathien (CIN) von hoher klinischer Relevanz. Die Prävalenz der CIN wird häufig mit 30–40 % angegeben, wobei je nach verwendeten Substanzen und Therapieregimen starke Abweichungen bestehen. Polyneuropathien treten auch aufgrund genetischer Ursachen und infolge von Vitaminmangel oder -überdosierung, Toxinen und Medikamenten sowie verschiedenen immunologischen Vorgängen auf. Viele Neuropathien sind behandelbar und sollten daher frühzeitig diagnostiziert werden. Etwa die Hälfte aller Polyneuropathien geht mit Schmerzen einher. Um neuropathischen Schmerz symptomatisch zu lindern, stehen medikamentöse Ansätze zur Verfügung. Körperliches Training sowie Physio- und Ergotherapie orientieren sich an individuellen Symptomen und funktionellen Defiziten.
Schlussfolgerung: Eine rasche Diagnose der Ursache für die Polyneuropathie ist entscheidend, um eine spezifische Therapie zu initiieren. Patienten mit schwerer Neuropathie unbekannter Ursache sollten zur gründlichen Diagnostik an spezialisierte Zentren überwiesen werden.
Polyneuropathien (PNP) sind generalisierte Erkrankungen des peripheren Nervensystems. Mit einer Prävalenz von circa 5–8 % sind sie hier die größte Erkrankungsgruppe (1). Aufgrund der zahlreichen Ursachen und Begleiterkrankungen kommen fast alle medizinischen Fachrichtungen mit Polyneuropathiepatienten in Kontakt.
Methodik
Dieser Artikel basiert auf einer selektiven Literaturrecherche in PubMed. Dabei wurden Publikationen aus den Jahren 2000–2017 zu den Suchbegriffen „neuropathy“, „polyneuropathy“, „diabetic neuropathy“,
„alcoholic neuropathy“, „chemotherapy induced neuropathy“, „chronic inflammatory demyelinating polyneuropathy“, „vasculitic neuropathy“ verwendet. Zusätzlich wurden aktuelle deutsche und europäische Leitlinien einbezogen. Die hereditären Neuropathien werden gesondert im Artikel von Eggermann et al. (e1) betrachtet.
„alcoholic neuropathy“, „chemotherapy induced neuropathy“, „chronic inflammatory demyelinating polyneuropathy“, „vasculitic neuropathy“ verwendet. Zusätzlich wurden aktuelle deutsche und europäische Leitlinien einbezogen. Die hereditären Neuropathien werden gesondert im Artikel von Eggermann et al. (e1) betrachtet.
Klinisches Bild und diagnostisches Vorgehen
Als häufigstes klinisches Bild tritt das distal symmetrische sensomotorische Syndrom auf. Abzugrenzen sind die Polyradikuloneuropathien mit proximalem und distalem Befall mit Rumpf- sowie Hirnnervenbeteiligung und die asymmetrische Mononeuropathia multiplex, bei der gleichzeitig oder versetzt unterschiedliche Nerven betroffen sind. Die klinischen Leitsymptome (Tabelle 1) (2) können diagnostisch wegweisend sein (Grafik). Je nach betroffenen Nervenfasertypen können sensible, motorische oder autonome Symptome im Vordergrund stehen, wobei jeweils Minussymptome wie Paresen oder Gefühlsstörungen und Plussymptome, zum Beispiel Faszikulationen, Muskelkrämpfe oder Schmerzen, unterschieden werden.
Tabelle 1
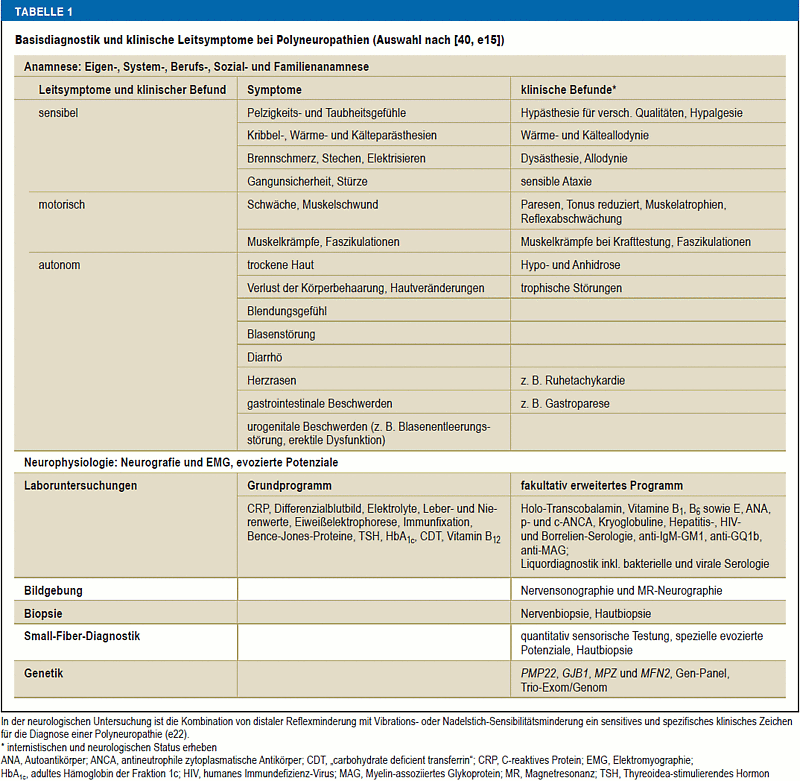
Basisdiagnostik und klinische Leitsymptome bei Polyneuropathien (Auswahl nach [40, e15])
Grafik
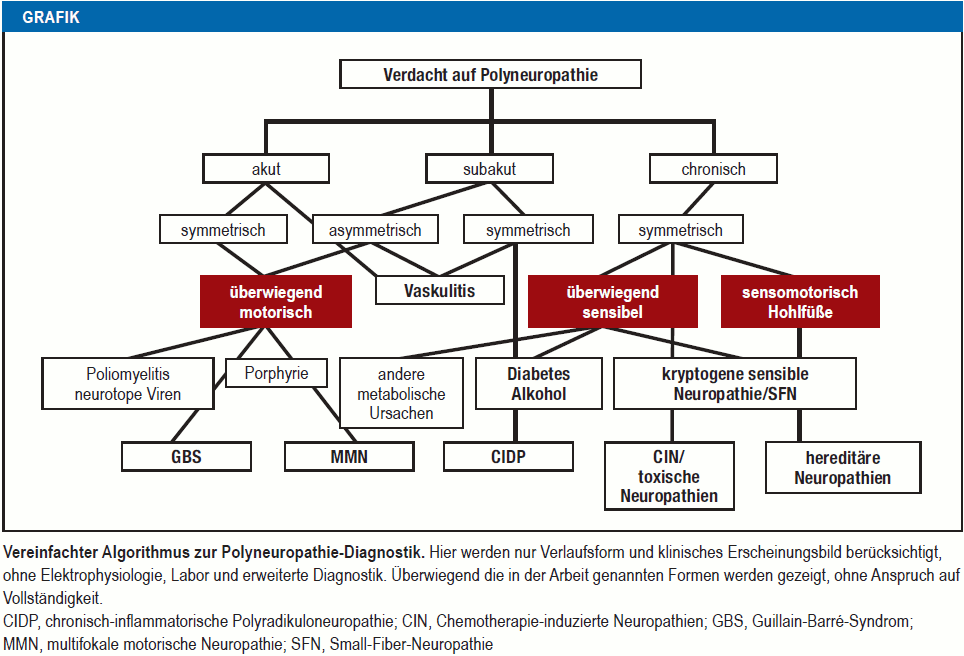
Vereinfachter Algorithmus zur Polyneuropathie-Diagnostik
Vorrangige Ziele der PNP-Diagnostik sind eine rasche Interventionsnotwendigkeit (Guillain-Barré-Syndrom, Vaskulitis) sowie behandelbare Ursachen (entzündlich, endokrinologisch, toxisch, nutritiv, tumorassoziiert) zuverlässig und rechtzeitig zu erkennen.
Ein wichtiger Parameter ist der Zeitverlauf, der sich von akut (zum Beispiel Guillain-Barré-Syndrom) über subakut (zum Beispiel Vaskulitis) zu chronisch (zum Beispiel Diabetes mellitus) bis hochchronisch (zum Beispiel hereditäre Neuropathien) spannt. Tabelle 1 fasst die Basisdiagnostik zusammen. Bei Verdacht auf eine entzündliche Genese ist eine Liquoruntersuchung erforderlich. Eine Indikation zur Nervenbiopsie besteht nur bei mittelschwer/schwer ausgeprägter progredienter Neuropathie, wenn weniger invasive Methoden nicht zur Diagnose geführt haben. Die Nervenbiopsie erlaubt eine Differenzierung zwischen demyelinisierender und axonaler Schädigung sowie den Nachweis von Entzündungszellen oder Amyloid (e2). Bei bis zu 30 % aller PNP bleibt die Ursache unklar. Ein Großteil hiervon sind kryptogene sensible PNP mit guter Prognose (e3). Bei Verdacht auf Small-Fiber-Neuropathie, einer PNP der dünnen Nervenfasern, deren Fehlfunktion der klinischen Elektrophysiologie entgeht, sind eine quantitative sensorische Testung (QST) und/oder Hautbiopsie indiziert. Auch hier sind Laboruntersuchungen erforderlich, um die Ursache zu klären (Tabelle 1) (3).
Pathophysiologie
Prinzipiell kann unterschieden werden zwischen Noxen, die primär die Nervenzelle, also das Motoneuron oder das Spinalganglienneuron, angreifen und solchen, die Prozesse in der Nervenfaser (Axon und Schwann-Zelle) stören (eGrafik). Letztere teilen sich auf in Affektionen der epi- und endoneuralen Blutgefäße (Vaskulitis, periphere arterielle Verschlusskrankheit [pAVK]), der Markscheiden sowie Ranvierschen Schnürringe (Demyelinisierung, Leitungsblockierung) und der Axone. Axonale Schädigungsmechanismen sind wiederum vielfältig, wobei Störungen des axonalen Transports von manchen Autoren als häufigste Schädigungsmechanismen bei erworbenen und hereditären Neuropathien angesehen werden (4).
eGrafik
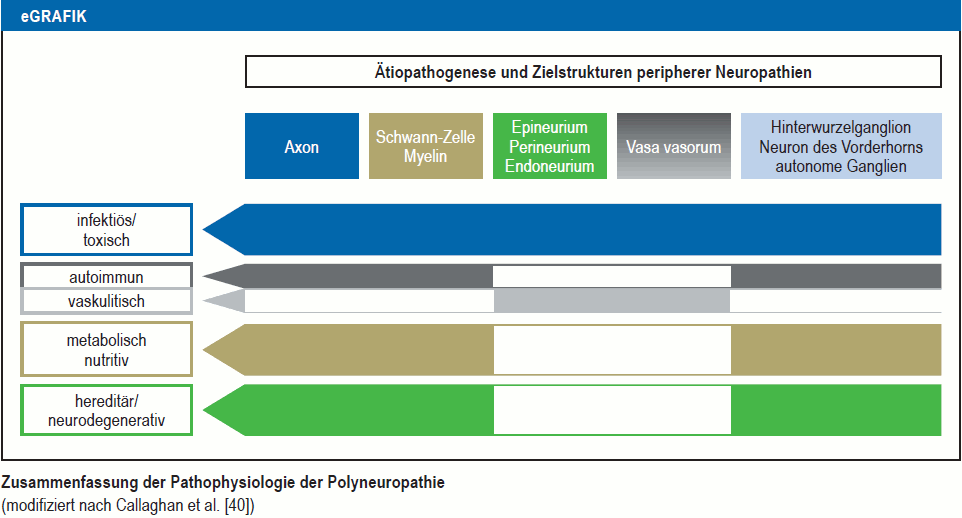
Zusammenfassung der Pathophysiologie der Polyneuropathie
Diabetische Neuropathie
Aufgrund der Pandemie von Prädiabetes und Diabetes ist die diabetische Neuropathie (DN) die häufigste PNP in Europa und wahrscheinlich weltweit (5). Laut der nationalen Versorgungsleitlinie für Diabetes im Erwachsenenalter beträgt die Prävalenz der DN 8–54 % bei Typ 1- beziehungsweise 13–46 % bei Typ 2-Diabetes (6). Die Ursache der PNP muss auch bei Verdacht auf DN abgeklärt werden, denn neben Diabetes können auch andere, gegebenenfalls behandelbare Ursachen vorliegen (e4). Zudem können auch zusätzliche Faktoren synergistisch dazu beitragen, dass die DN fortschreitet (Kasten 1) (5–7). Pathophysiologisch spielen folgende multiple Faktoren zusammen:
- Störungen der Mikrozirkulation
- Beeinträchtigung des Mitochondrien- und Fettstoffwechsels
- Aktivierung alternativer Stoffwechselwege
- Bildung neurotoxischer glykierter Proteine (5).
Kasten 1

Risikofaktoren für die Entwicklung einer diabetischen Polyneuropathie (7)
Die häufigste Form ist die distal symmetrische PNP, die mit sensiblen Symptomen (Taubheitsgefühl, Parästhesien) oder auch als Small-Fiber-Neuropathie (Schmerzen, Verlust der Temperaturempfindung) beginnt (e5). Sollten klinisches Bild und Elektroneurographien nicht zur Diagnose führen, empfiehlt sich die QST, gegebenenfalls eine Hautstanzbiopsie oder eine Funktionsdiagnostik autonomer Nervenfasern (6). Motorische Ausfälle treten bei der distal symmetrischen DN allenfalls spät im Erkrankungsverlauf auf, stehen jedoch bei anderen Formen im Vordergrund. Dies sind Mononeuropathien wie Okulomotoriusparese oder die diabetische Amyotrophie/Plexopathie. Selten treten akute schmerzhafte und autonome Neuropathien, die früher Insulinneuritis genannt wurden, zu Beginn intensiver Insulin-Therapien auf (8). In Anbetracht der Vielfalt dieser Neuropathieformen gibt es nicht „die Therapie“ der DN. Laut nationaler Versorgungsleitlinie sollen Patienten bei allen Formen und in allen Stadien der DN in Bezug auf Lebensgewohnheiten, Diabeteseinstellung und Fußpflege beraten werden (6). Sowohl bei Patienten mit Typ 1- als auch bei Typ 2-Diabetes soll eine individuell und dem Komorbiditätsprofil angepasste Diabeteseinstellung erfolgen. Die Therapie setzt sich aus folgenden Elementen zusammen:
- Kontrolle der zusätzlichen Risikofaktoren
(Kasten 1) - Umstellung des Lebensstils inklusive körperlichem Training
- symptombezogene Behandlung, zum Beispiel Schmerztherapie, vegetative Störung oder diabetisches Fußsyndrom.
Ob Immuntherapien bei der diabetischen Amyotrophie wirksam sind, muss weiter untersucht werden (9).
Alkohol-assoziierte Polyneuropathie
Die Alkohol-assoziierte PNP hat eine Prävalenz von 22–66 % unter chronischen Alkoholikern. Die wichtigsten Faktoren sind die Dauer des Missbrauchs und die Lebenszeit-Alkoholmenge. Spiegeltrinker sind stärker betroffen als episodische Trinker, Frauen stärker als Männer (10). Eine Menge von > 100 g/Tag über mehrere Jahre gilt als wahrscheinlich pathogen für eine PNP (e6). Die Pathophysiologie der Alkohol-assoziierten PNP setzt sich zusammen aus Mangelernährung, zum Beispiel im Hinblick auf B-Vitamine, und direkten toxischen Einflüssen von Alkohol sowie seinen Abbauprodukten wie Acetaldehyde. Oxidativer Stress spielt ebenfalls eine Rolle. In der Regel sind die Leberwerte und die Konzentration an „carbohydrate deficient transferrin“ (CDT) erhöht. Zudem liegt meist eine Makrozytose vor. Anfangs bestehen Störungen der Sensibilität mit und ohne neuropathische Schmerzen. Später können distal betonte Paresen und vegetative Funktionsstörungen dazukommen. Neurografisch zeigt sich eine axonale sensomotorische Neuropathie. Neuropathologisch sind vor allem die dünnen Nervenfasern betroffen (e7), was die Schmerzhaftigkeit erklärt. Die Therapie umfasst eine Alkoholabstinenz und eine Umstellung des Essverhaltens, um die Mangelernährung zu korrigieren. Bei eingehaltener Abstinenz kann sich die Neuropathie innerhalb von Monaten bis Jahren zurückbilden (11, e8
Chemotherapie-induzierte Neuropathie und
andere toxische Neuropathien
Die Chemotherapie-induzierte Neuropathie (CIN) ist die häufigste neurologische Nebenwirkung einer Tumortherapie mit Zytostatika wie Platinderivaten, Vinca-Alkaloiden, Taxanen, Proteasomen-Inhibitoren, aber auch moderner Antikörper-basierter Therapien. Aufgrund der Zunahme von Tumorerkrankungen und höheren Langzeitüberlebensraten steigt die Inzidenz der CIN. Die Zahlen variieren in Abhängigkeit der verwendeten Substanzen und Regime sowie der Art des Assessments, häufig werden 10–90 % oder 30–40 % angegeben (12). Typischerweise beginnt die CIN mit sensiblen Ausfallsymptomen sowie Schmerzen innerhalb der ersten 2 Monate der Therapie und kann sich stabilisieren beziehungsweise zurückbilden, nachdem die Therapie abgesetzt wurde (12). Während beispielsweise im Fall von Oxaliplatin akute neurotoxische Phänomene bei 60–80 % der Patienten innerhalb von 2–3 Tagen nach Applikation reversibel sind, entwickeln sich bei 73 % mit zunehmender Therapiedauer persistierende Strukturschäden an Spinalganglien und peripheren Nerven (13). Platin-, seltener auch Vincristin-basierte Therapien können zum „Coasting“-Phänomen führen, einer zunächst weiteren Verschlimmerung nach Absetzen der Substanz (12). Etwa 40 % der CIN gehen mit chronischen Schmerzen einher, wobei eine neuropathische und eine vermutlich sekundäre, auf muskulärer Fehlfunktion beruhende, myofasziale Komponente bestehen können (12, 14, e9). Im Falle des Proteosomen-Inhibitors Bortezumib tritt vorwiegend eine Small-Fiber-Neuropathie auf. Neuere onkologische Therapieansätze mit immunmodulatorischen Antikörpern, den sogenannten Checkpoint-Inhibitoren mit den Zielstrukturen „cytotoxic T-lymphocyte-associated protein 4“ (CTLA-4) oder „programmed cell death protein 1“ (PD-1)-Rezeptor, können akute und chronische Immunneuropathien induzieren. Sie werden durch Absetzen des verursachenden Medikaments und nach den Regeln für die Therapie von Immunneuropathien behandelt (12).
Die Neurotoxizität ist abhängig von der Höhe der Einzeldosis, der kumulativen Gesamtdosis sowie der Chemotherapiedauer. Ein engmaschiges klinisches Monitoring mit Anamnese der PNP-Symptome und klinisch-neurologischer Untersuchung ist erforderlich, um Dosis, Therapieintervalle oder -regime anpassen zu können. Schweregrad und Beeinträchtigung der Lebensqualität können standardisiert erfasst werden (e10, e11). Präventiv könnte die Identifizierung von genetischen Risikofaktoren für spezifische Zytostatika, zum Beispiel Platin oder Vincristin, relevant werden (12, 13, 15).
Sonstige toxische Neuropathien
Zahlreiche Medikamente und Umweltgifte können eine Polyneuropathie auslösen (Kasten 2). Das Vermeiden der Exposition und die rasche Elimination der Toxine stehen im Vordergrund der Therapie. Bei Schwermetallen kann die Elimination durch Komplexbildner sowie forcierte Diurese gefördert werden (16).
Kasten 2

Auslöser toxischer Polyneuropathien (Auswahl)
Neuropathien bei Vitaminmangel
und Vitaminüberdosierung
und Vitaminüberdosierung
Bei Vitamin-B12-Mangel kann ein subakutes Beschwerdebild mit Kribbelparästhesien der Füße, sensibler Ataxie und Hypästhesie auftreten. Paresen kommen selten vor. Unbehandelt können sich eine Optikusatrophie, eine Depression oder eine Demenz entwickeln (17). Sind die Aβ-Fasern und die Hinterstränge des Rückenmarks beteiligt, liegt eine kombinierte Myeloneuropathie vor. Dies bedingt gesteigerte Muskeldehnungsreflexe und positive Pyramidenbahnzeichen, woran diese Form der PNP leichter erkennbar ist. Bei etwa der Hälfte der Patienten mit neurologischen Symptomen ist die für Vitamin-B12-Mangel typische makrozytäre Anämie nicht nachweisbar. Eine Vitamin-B12-Substitution sollte so rasch wie möglich begonnen werden. Ein Vitamin-B6-Mangel kann zur subakuten sensomotorischen PNP führen. Mehrere Fälle wurden als Komplikation der Behandlung von Morbus Parkinson mit intestinalen Duodopa-Pumpen (18) und nach rascher Gewichtsabnahme beschrieben. Da auch eine Überdosis von Vitamin B6 zu einer PNP führen kann, solle eine unkontrollierte Einnahmen vermieden werden (19).
Immunneuropathien
Guillain-Barré-Syndrom
Ein Patient mit Zustand nach gastrointestinalem oder respiratorischem Infekt, der nun über aufsteigende Lähmungen klagt, hat mit hoher Wahrscheinlichkeit ein Guillain-Barré-Syndrom (GBS), was eine rasche Klinikeinweisung und oft intensivmedizinische Behandlung notwendig macht. Bei dieser akuten Polyradikuloneuritis mit einer Inzidenz von 1–2/100 000 Fälle pro Jahr können neben der rasch aufsteigenden Tetraparese bedrohliche kardiale Erregungsleitungsstörungen auftreten und auch die Atemmuskulatur versagen. Die Therapie dieser autoimmunen Neuropathie besteht in engmaschigen Kontrollen, supportiven Maßnahmen und der Gabe von intravenösen Immunglobulinen (IVIg) oder Plasmapheresen (20) (eTabelle).
eTabelle
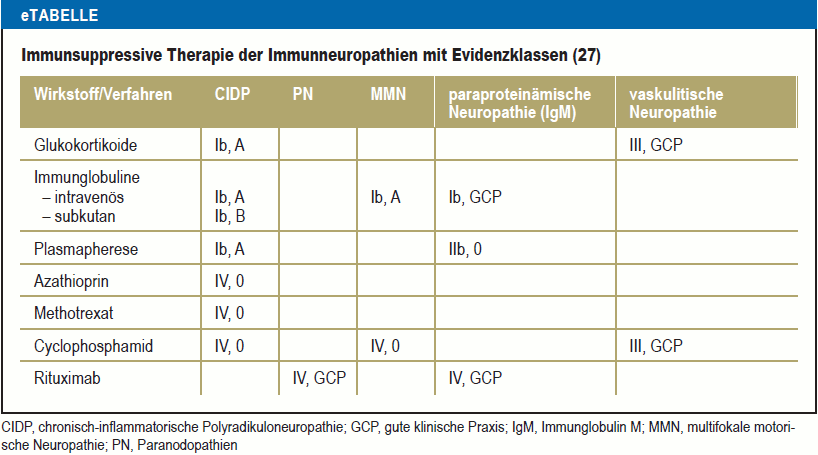
Immunsuppressive Therapie der Immunneuropathien mit Evidenzklassen (27)
Chronisch-inflammatorische Polyradikuloneuropathie
Im Gegensatz zum GBS, das definitionsgemäß seinen Nadir nach 4 Wochen erreicht, ist die chronisch-inflammatorische Polyradikuloneuropathie (CIDP) eine chronische Autoimmunerkrankung mit einer Entwicklung über mindestens 8 Wochen. Die Prävalenz beträgt 2–3/100 000 Fälle. Das klinische Bild besteht in einer symmetrischen, motorisch betonten Polyradikuloneuropathie mit distaler wie proximaler Muskelschwäche, Areflexie, Parästhesien und sensiblen Defiziten. Der Verlauf ist zumeist chronisch progredient, seltener schubförmig remittierend. Es gibt Formen mit asymmetrischer Verteilung, rein motorischen oder rein sensiblen Störungen (21). Die Diagnose stützt sich auf das klinische Bild, elektrophysiologische Zeichen der Demyelinisierung, nicht obligate Liquorkriterien mit weniger als 10 Leukozyten/μL sowie Liquor-Eiweißerhöhung und gegebenenfalls den Nachweis der Demyelinisierung mittels Suralisbiopsie (21). In den letzten Jahren haben zusätzlich Ultraschall und Magnetresonanztomographie (MRT) der Nerven die Diagnostik und das Therapiemonitoring verbessert (e12).
Evidenz aus kontrollierten Studien liegt für die Wirksamkeit von Glukokortikoiden (GC), IVIg und die Plasmapherese vor (22). Die Plasmapherese wird wegen hohen Aufwands und kurzer Wirkdauer überwiegend bei akuten Verschlechterungen angewendet (23). Ob in der Langzeittherapie GC oder IVIg günstiger sind, ist noch offen. Initial sprechen mehr Patienten auf IVIg an, allerdings scheinen die GC-Responder nach Absetzen der Therapie länger in Remission zu gehen (e13). Nach Expertenmeinung wird die Entscheidung über IVIg oder GC bei CIDP in Abwägung der zu erwartenden Nebenwirkungen und Kosten getroffen (24). Wenn GC gegeben werden, wird die Pulstherapie gegenüber der oralen Dauertherapie bevorzugt (25). Laut Studienlage ist eine subkutane Immunglobulin-Therapie wirksam (26). Noch ist allerdings keines der Präparate für diese Indikation zugelassen. Bei keiner der anderen Substanzen für eine Langzeitimmuntherapie wie Azathioprin, Methotrexat oder Interferon beta-1a wurde eine Wirkung bei CIDP in randomisierten kontrollierten Studien nachgewiesen (27, 28). Die Evidenzlagen finden sich in der eTabelle.
Paraproteinämische Neuropathien
Unter diesem Begriff werden alle PNP zusammengefasst, bei denen im Serum der Patienten ein Paraprotein gefunden wird. Allerdings ist dies in Anbetracht der Häufigkeit von Paraproteinen, aber auch von PNP im höheren Lebensalter meist ein zufälliges Zusammentreffen, das keine Konsequenzen für die Behandlung der PNP hat. Hiervon ausgenommen sind folgende Konstellationen:
- Eine meist distal und motorisch betonte Polyneuropathie mit ausgeprägten Zeichen einer Demyelinisierung (29) tritt bei IgM-Gammopathie, oft mit Antikörperreaktivität gegen Myelin-assoziiertes Glycoprotein auf. Die Therapie erfolgt zunächst analog zur CIDP, wobei einige Autoren den Einsatz von Rituximab empfehlen (30).
- Bei PNP und IgGλ-Paraproteinämie mit angiofollikulärer Lymphknotenhyperplasie (Morbus Castleman), osteosklerotischen Knochenläsionen oder erhöhtem vaskulären endothelialen Wachstumsfaktor kann ein POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, M-Protein, „skin changes“) vorliegen. Die Behandlung der ersten Wahl ist eine autologe Stammzelltransplantation (31).
- Bei großen Mengen des Paraproteins muss an eine hämatologische Erkrankung gedacht werden. Ein hoher IgM-Wert kann auf Morbus Waldenstrom, eine gestiegene IgG-Konzentration auf eine AL-Amyloidose beim Myelom hinweisen. Die Evidenzlagen finden sich in der eTabelle.
Paranodopathien
Nachdem Autoantikörper gegen paranodale Proteine am Ranvierschen Schnürring wie Neurofascin-155, Contactin-1 und Caspr-1 bei Patienten mit dem klinischen Bild einer CIDP entdeckt wurden, entstand der Begriff der Paranodopathien. Typisch ist ein akuter Beginn wie bei GBS mit Übergang in einen chronischen Krankheitsverlauf. Klinisch besteht eine akute schwere motorisch betonte, elektrophysiologisch meist axonale Neuropathie, oft mit Aktionstremor und Ataxie. Bei dieser meist IgG4-assoziierten Immunneuropathie wird Rituximab als Therapie der 1. Wahl angesehen, wobei die Patienten initial auch auf die klassische CIDP-Therapie ansprechen (32, e14, e15).
Multifokale motorische Neuropathie
Die multifokale motorische Neuropathie (MMN) hat eine Prävalenz von 0,6–2 pro 100 000 Fälle. Diese rein motorische Neuropathie ist durch progrediente, distal betonte und asymmetrische Paresen sowie Atrophien gekennzeichnet. Sie betrifft Männer bevorzugt und startet meist an den oberen Extremitäten. Typisch ist der Nachweis multifokaler Leitungsblöcke in den motorischen Neurographien, unabhängig von physiologischen Engstellen. Bei circa 50 % der Betroffenen können serologisch hochtitrige IgM-Antikörper gegen die Ganglioside GM1 nachgewiesen werden (33). Therapie der Wahl ist die repetitive Gabe von IVIg. Die subkutane Applikation von Immunglobulinen ist wirksam, jedoch noch nicht zugelassen (34). Andere Immunsuppressiva einschließlich GC sind wirkungslos (27, e16). Die Evidenzlagen finden sich in der eTabelle.
Vaskulitische Neuropathien
Bei progredientem multifokalen Befall verschiedener peripherer Nerven, aber auch bei subakuter distal symmetrischer PNP muss an eine Vaskulitis als Ursache gedacht werden (eAbbildung). Manchmal manifestiert sich eine systemische Vaskulitis primär als PNP, zum Beispiel bei mikrovaskulärer Polyangiitis oder bei eosinophiler Granulomatose mit Polyangiitis. Häufig liegt jedoch eine isolierte Vaskulitis des peripheren Nervensystems vor (35), sodass die Diagnose nur mittels Nervenbiopsie möglich ist. Eine kombinierte Nerv-Muskel-Haut-Biopsie erhöht die Trefferrate (e17, e18). Die Behandlung erfolgt mit GC. Bei Therapieresistenz werden Cyclophosphamid oder Rituximab eingesetzt, analog zu systemischen Vaskulitiden (27) (Evidenzlagen in der eTabelle).
eAbblidung
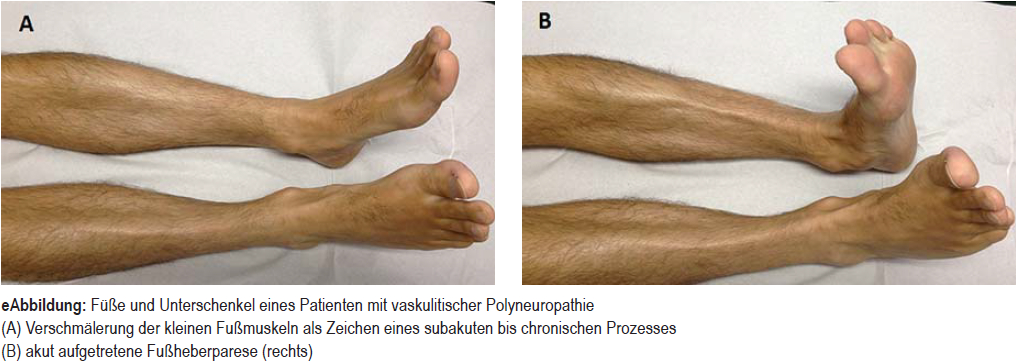
Füße und Unterschenkel eines Patienten mit vaskulitischer Polyneuropathie
Symptomatische Therapie
Therapie des neuropathischen Schmerzes
Etwa die Hälfte aller Polyneuropathien geht mit Schmerzen einher (35, e19, e20). Diese neuropathischen Schmerzen entstehen, vereinfacht beschrieben, durch Spontanaktivität und Sensibilisierung geschädigter Axone, vermittelt durch überaktive Natrium-Kanäle sowie die Einwirkung von Entzündungsmediatoren und Wachstumsfaktoren. Durch permanenten Einstrom nozizeptiver Information ins Rückenmark und Gehirn kann dort das Phänomen der zentralen Sensibilisierung auftreten – zusätzlich zum Versagen der tonischen und phasischen endogenen Schmerzhemmung (e21). Da die Mechanismen neuropathischer Schmerzen sich somit grundlegend von denen nozizeptiver Schmerzen unterscheiden, sind spezielle Therapieansätze notwendig (36). Die medikamentöse Therapie neuropathischer Schmerzen wurde kürzlich in einer Metaanalyse mit Empfehlungen zusammengefasst (36). Medikamente der ersten Wahl sind Gabapentin, Pregabalin, Duloxetin und Trizyklika, wobei differenzielle Indikationen sowie das Nebenwirkungsprofil zu beachten sind (Tabelle 2). Topische Therapien wie Lidocain- oder Capsaicin-Pflaster können bei umschriebenen Schmerzarealen hilfreich sein (37, 38)
Tabelle 2
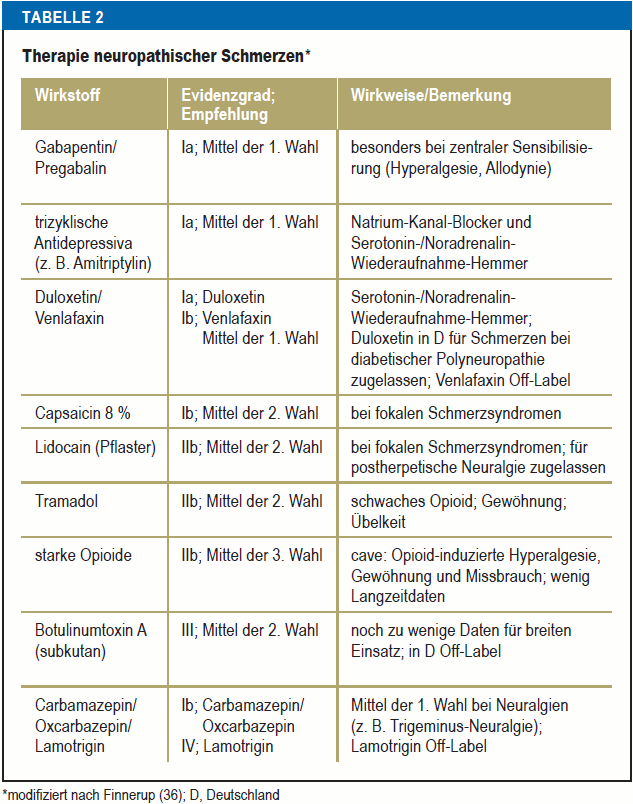
Therapie neuropathischer Schmerzen*
Physio-, Ergo- und Trainingstherapie
Die Physiotherapie bei Neuropathien orientiert sich an Symptomen und funktionellen Defiziten. Sie beinhaltet Übungen, die Stand- und Gangsicherheit verbessern und Gleichgewicht, Koordination sowie Propriozeption schulen sollen. Bei Paresen wird angestrebt, die Muskelkraft sowie -funktion zu erhöhen und die muskuläre Balance als Prävention von Deformitäten sowie Kontrakturen zu erhalten oder wiederherzustellen. Physikalische und balneologische Therapiemaßnahmen können zusätzlich eingesetzt werden. Bei Beeinträchtigung der Handfunktion ist Ergotherapie, gegebenenfalls ergänzt durch entsprechende Hilfsmittelanpassung, indiziert. Sportliche Aktivität ist im Rahmen der erhaltenen Funktion erwünscht. Bei vielen Patienten sind die proximalen Muskelgruppen lange Zeit kaum betroffen und somit trainierbar. Bei CIDP wirkt sich dreimal wöchentliches Ergometer- und Krafttraining positiv auf Ausdauer sowie Muskelkraft aus (39).
Mehr als jeder zehnte Multiple Sklerose-Erkrankte ist von Augenbewegungsstörungen betroffen
von MS-Register
Quelle: msfp, DMSG-Bundesverband - 07. 12. 2018
Redaktion: DMSG Bundesverband e.V.
aus https://www.dmsg.de/multiple-sklerose-news/ms-forschung/news-article/News/detail/mehr-als-jeder-zehnte-multiple-sklerose-erkrankte-ist-von-augenbewegungsstoerungen-betroffen/?cHash=d7894f098f3ba0ee749c770f8bebddad&L=0
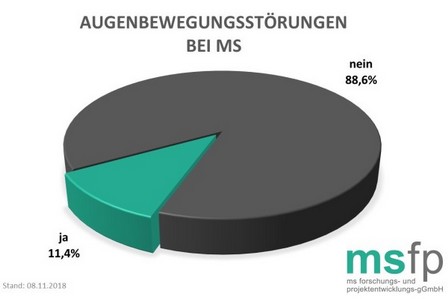
Doppelbilder, verschwommenes Sehen, Gleichgewichtsstörungen: 11,4 Prozent der befragten MS-Erkrankten in Deutschland gaben zum Zeitpunkt der letzten Meldung für das MS-Register der Deutschen Multiple Sklerose Gesellschaft, Bundesverband e.V. an, unter MS-bedingten Augenbewegungsstörungen (Okulomotorische Störungen) zu leiden.
Dieses MS-Symptom, das durch Entzündungen verschiedener Hirnnerven und/oder Wärme (Uhthoff-Phänomen) hervorgerufen bzw. verstärkt werden kann, führt dazu, dass die Bewegungen der Augäpfel nicht mehr richtig gesteuert werden. Dies äußert sich beispielsweise in Doppelbildern, verschwommenem Sehen, Gleichgewichtsstörungen oder auch in einem Augenzittern (Nystagmus). Eine medikamentöse Behandlung der MS-bedingten Bewegungsstörungen der Augen ist möglich, führt allerdings nicht immer zum gewünschten Erfolg. Durch Wärme ausgelöste Augenbewegungsstörungen können durch abkühlende Maßnahmen gemildert werden.
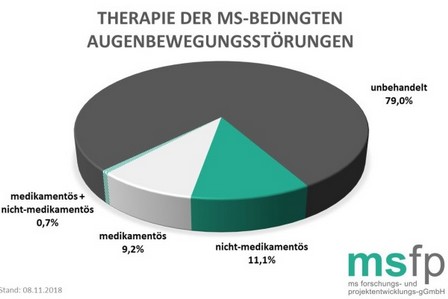
Wie verläuft die Therapie?
Augenbewegungsstörungen bleiben bei über drei Viertel der befragten MS-Erkrankten (79,0 Prozent) unbehandelt. Innerhalb der 21,0 Prozent Patienten, die behandelt werden, erhalten 53,0 Prozent eine nicht-medikamentöse Therapie, 43,8 Prozent erhalten eine medikamentöse Behandlung und weitere 3,2 Prozent eine Kombination aus medikamentöser und nicht-medikamentöser Therapie. Die am häufigsten angewendete nicht-medikamentöse Behandlung der MS-bedingten Augenbewegungsstörungen ist mit 27,6 Prozent der Einsatz von Hilfsmitteln, gefolgt von der Physiotherapie mit 15,5 Prozent und der Ergotherapie mit 13,1 Prozent.
Über das MS-Register
Schätzungen zufolge leben in Deutschland mehr als 240.000 MS-Erkrankte. Verlässliche Daten zur Prävalenz (Häufigkeit) der Erkrankung lagen lange Zeit nicht vor. Auch Fragen zur Versorgung, wie zum Beispiel die Anwendung verschiedener Therapieformen bei MS-Patienten waren nur ansatzweise bekannt. Um standardisierte Daten über die Versorgungssituation innerhalb Deutschlands und den Einfluss der Erkrankung auf die Berufs- und Arbeitswelt zu erhalten, initiierte die Deutsche Multiple Sklerose Gesellschaft, Bundesverband e.V. im Jahre 2001 den Aufbau eines MS-Registers. Mittlerweile beteiligen sich über 180 MS-Zentren an der Dokumentation.
Generationenprojekt auf VOX zeigt, dass die körperliche und geistige Leistungsfähigkeit von Senioren gesteigert werden können. Stetig neue Herausforderungen fördern die Neuroplastizität der Senioren.
Das Resultat ist erstaunlich...