Newsletter
Aktuelle Newsletter
Viel Spaß beim Lesen!
Klonus- Symptom oder Krankheit?
Chantal Ungruhe - neku
Wer kennt es nicht, die Behandlung des Klienten ist beende, man stellt die Füße zurück auf die vorgesehene Fußraste vom Rollstuhl und der Fuß beginnt zu zucken. Was nun? Runterdrücken und warten? Oder den Fuß wieder auf den Boden stellen und aus der Situation nehmen? Oder einfach abwarten und hoffen das es vorbei geht?
Was ist ein Klonus? Wie sieht die Therapie dafür aus und was für Möglichkeiten habe ich, wenn ein Klonus aktiv ist. Über diese Fragen werden wir einen Überblick schaffen und versuchen etwas Klarheit zu entwickeln.
Fangen wir aber mal von vorne an:
Was ist ein Klonus überhaupt?
Einfach übersetzt bedeutet Klonus „heftige Bewegung“ (1). Es ist ein oszillierender Dehnungsreflex, wo die Ursache der Entstehung immer noch nicht ganz geklärt ist. (2)
Es sind unwillkürliche Bewegungen der Muskelfasern, welche sich schnell abwechselnd An- und wieder Entspannen. Diese Muskelzuckungen sind die Antwort auf eine Endstellung vom Gelenk, wobei die Sehnen schnell und abrupt, passiv gedehnt werden. (3) Zurück auf das Beispiel mit der Fußraste vom Anfang, ist es doch so, dass bei vermehrter Dorsalextension des Fußes auf der Stütze, die Achillessehne mehr gedehnt wird und so eine passive Dehnung dieser Sehne entsteht. Dadurch wird der Klonus ausgelöst und das rhythmische Zucken beginnt.
Ein Klonus tritt mit am häufigsten in den Extremitäten, vor allem aber im Fuß oder Kniegelenk auf, also der Achillessehnenreflex und der Patellarsehnenreflex (3). Seltener kann er zusätzlich noch in folgenden Bereichen auftreten:
- Handgelenke
- Finger
- Kiefer
- Ellenbogen
- Bizeps
Symptom oder Krankheit?
Um dies zu beantworten, fangen wir mal ganz oben an, beim 1. Motorischen Neuron im Gehirn. Hierbei handelt es sich um eine Nervenbahn im ZNS, welche in der Großhirnrinde entspringt und mit seinem Axon die Signale für die Muskulatur, um eine bewusste Bewegung (Willkürmotorik) auszulösen, an das 2. Motorische Neuron weiterleitet, welches dann die Signale an die Muskulatur weiterleitet und mit dem Muskel verbunden ist, damit die Signale ankommen (4).
Bei einer Läsion oder Schädigung des 1. Motorischen Neurons oder auch Upper Motor Neuron Syndrom genannt, gibt es verschiedene Symptome, die auf eine Schädigung des UMNS hinweisen. Hier muss man jedoch nochmal unterscheiden, da je nach geschädigter Struktur eben die Pyramidenbahnen oder auch extrapyramidale Bahnen betroffen seien können (5).
Pyramidenbahnzeichen sind Pathologische Reflexe, die nochmal in Plus und Minus Symptome eingeteilt werden. Hierbei ist die Kontrolle durch die Fasern der Pyramidenbahn defekt oder gestört, sodass nicht nur eine kurze Erregung des Reflexes stattfindet, sondern eine konstante Dauererregung.
Plus oder Positiv Symptome bedeutet nicht, dass es gute Symptome sind, sondern man redet hier von einer verstärkten Aktivität oder von zu viel, also als Beispiel: zu viel Tonus.
Genauso ist es mit Minus oder Negative Symptome gemeint. Hier bedeutet es, zu wenig oder kaum Aktivität.
Merkmale der Plussymptome sind:
- Spastisch erhöhter Muskeltonus
- Pathologische Reflexe (Z.B. Babinski)
- Gesteigerte Muskeleigenreflexe
- Klonus
- Assoziierte Reaktionen
- Bewegungssynergie
Merkmale der Negativsymptome sind dementsprechend das Gegenteil:
- Parese
- Muskelschwäche
- Ermüdung, Langsamkeit
Ein positiver Babinski ist das Hauptmerkmal jeder Pyramidenbahnstörung, es ist jedoch nicht auszuschließen das eine Pyradmidenbahnstörung vorliegen kann, obwohl der Babinski negativ ist (6). Beim Babinski-Reflex wird am äußeren Fußrand entlang gestrichen und erwartet, dass bei einem positiven Zeichen, eine Dorsalextension der Großzehe bei einer gleichzeitigen Spreizung der übrigen Zehen entsteht.
Nun aber zurück zum Klonus. Es ist ebenfalls ein Bestandteil der Plussymptomatik und deutet somit auf eine Beschädigte Nervenbahn im Hirn. Man kann davon ausgehen das bei diversen Erkrankungen im Hirn ein Klonus zu erwarten ist.
Besonders häufig tritt es bei Multipler Sklerose, Schlaganfall, Infektionen (Meningitis) aber auch bei einem Hirntumor oder einer Zerebralparese auf (7).
Man vermutet, dass durch die beschädigten Nervenbahnen oder auch Pyramidenbahnläsion, ein Mechanismus ausgelöst wird, der eine konstante Aktivierung der Muskeldehnungskreise bedingt. Also jede neue Muskelzuckung führt wieder dazu, dass der Reflex nochmal aktiviert wird, sodass ein Teufelskreis entsteht, der niemals enden wird, solange die Position der Extremität gehalten wird (8). Die Impulse der afferenten Bahnen (also zum Muskel führend) haben eine fehlende Hemmung auf die Alpha-Motoneurone, welche für die Muskelkontraktion zuständig sind (9).
Klonus ist eine langfristige Erkrankung, wo die Behandlung aufgrund der verschiedenen Ursachen variiert. Wichtig ist herauszufinden, was den Patienten*innen am besten hilft und das diverse Möglichkeiten zur Verfügung stehen, um eine Verbesserung oder Linderung zu erreichen. Da dieser Mechanismus aber den Reiz immer wieder neu auslöst, können wir schlecht diesen Reiz einfach hemmen. Es müssen also andere Reize gesetzt und gehemmt werden, sodass die Patient*innen Strategien entwickeln können, den Klonus aufzulösen. Je früher man mit Physiotherapie anfängt, desto besser.
Am einfachsten ist es, wenn eine andere Position gesucht wird, wo der Reiz gehemmt wird. Als Beispiel beim Beintremor: Hier können Wir versuchen, durch das Anheben des Beines, die Hand in die Kniekehle zu legen und so eine Abschwächung des Reizes zu erreichen. Physiotherapie kann den Patient*innen helfen, um verschiedene Strategien zu erarbeiten, damit Sie besser mit dem Klonus leben können. Es kann massiv den Alltag einschränken, da die Muskulatur durch die ständigen Kontraktionen schwächer und müde wird und somit die Patient*innen eine Schwächere Muskulatur haben, schneller ermüdbar sind und für einige Aufgaben ihre Energie schon verbraucht haben.
Ebenso kann Ausdauersport mit seinen gleichmäßigen Muskelbewegungen auch dazu beitragen, die Reizüberflutung abzuschwächen. Patient*innen können wunderbar bei niedriger Belastungsstufe Fahrradfahren. Am besten jeden Tag!
Eine weitere Möglichkeit zur Behandlung sind Medikamente. Es ist jedoch so, dass es sich hierbei hauptsächlich um Antispastika und Beruhigungsmittel handelt, welche nur global auf den Klonus einwirken und ebenfalls mit starken Nebenwirkungen einhergehen. Sie machen müde und träge, oder sie können auch Vagilanzstörungen führen. Sie ändern auch an dem Grundproblem, der ständigen Reizauslösung nichts. Sie hemmen das System, lösen aber nicht die Ursache vom Klonus.
Andere Therapien können Botox-Injektionen sein, Entspannungsübungen oder in Physiotherapie kann mit Wärme oder Kälte gearbeitet werden, um so zu versuchen die Schmerzen zu lindern. Dehnungsübungen sind ein wichtiger Bestandteil der Therapie, ebenso kann eine Schienenversorgung manchen Patienten helfen. Als letzter Ausweg kann eine Operation eine Möglichkeit sein. Hier werden die Nervengänge durchtrennt, die eben zu den unwillkürlichen Muskelzuckungen führen. (10)
Klonustest
Der Klonustest gehört zu einem festen Bestandteil einer neurologischen Untersuchung, um das Nervensystem bewerten zu können.
Der Test kann einfach und leicht durchgeführt und getestet werden ohne irgendwelches Kosten oder Materialaufwand. Zur Erläuterung ist es hier am Beispiel eines Fußklonus erläutert, der aber auch leicht abgewandelt werden kann auf die Hand.
Beim dem Test sitzen die Patient*innen mit hängenden lockeren Unterschenkel auf der Bank ohne Fußsohlenkontakt. Nun umgreift der Tester mit einer Hand den Vorfuß und mit der anderen fixiert er den Unterschenkel. Jetzt bringt der Tester den Vorfuß ruckartig in eine Dorsalextesion im OSG und hält diese Position. Positiv ist der Test, wenn eine kontinuierliche rhythmische Kontraktion der Wadenmuskulatur zu sehen und spüren ist. (11)
Je nach Dauer der Muskelkontraktion unterscheidet man in 2 Klonusarten: Den erschöpflichen und unerschöpflichen Klonus. Es bezieht sich aber nicht auf die Dauer des Klonus, sondern auf die Reaktion des Auslösers.
Der erschöpfliche Klonus lässt sich teilweise sogar bei einem gesunden Muskel auslösen. Er hat, im Gegensatz zum unerschöpflichen Klonus, eine weniger werdende Amplitude und hört nach kurzer Zeit ganz auf, sobald man den Muskel in Ruhe lässt. Wenn ich einen erschöpflichen Klonus auf beiden Seiten finde, ist es sogar physiologisch und mein Patient hat einfach ein sehr lebhaftes Reflexniveau. Tritt er aber nur an einer Seite auf oder ist sogar unerschöpflich, könnte er auf eine Pyramidenbahnläsion hinweisen.(12)
Beim unerschöpflichen Klonus ist es so, dass dieser bestehen bleibt, wenn man den Muskel dauerhaft unter Stress setzt, also eine Überdehnung, und die Amplitude nicht weniger wird sonst das Zucken weiterhin besteht bei gleicher Position. Dieser Klonus ist fast immer pathologisch zu sehen und ein Zeichen für eine Pyramidenbahnläsion. (13)
Zusammengefasst ist es wichtig zu wissen, dass ein Klonus ein Symptom des Pyramidenbahnzeichen ist und es therapierbar ist. Es gibt diverse Möglichkeiten und die gilt es auch zu nutzen.
Literatur
1) Klonus. (2017, 4. September). online-heilpraktikerschule-mv.de. http://online-heilpraktikerschule-mv.de/klonus/ Aufgerufen am 12.2.23 um 11:50
2) Zimmerman B, Hubbard JB. Clonus. 2022 Aug 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 30521283.
3) Engelhardt Lexikon Orthopädie und Unfallchirurgie. (o. D.). https://www.lexikon-orthopaedie.com/pdx.pl?dv=0 Aufgerufen am 12.2.23 um 12:03
4) Füeßl, H. S. (2017, 7. Februar). Was bedeutet ein Klonus beim Auslösen der Reflexe? SpringerLink. https://link.springer.com/article/10.1007/s15006-017-9211-7?error=cookies_not_supported&code=37a50c77-c579-4d83-9c70-e254c1079e7fAufgerufen am 12.2.23 um 12:31
5) GmbH, D. C. (o. D.). Motoneuron Aufgerufen am 12.2.23. DocCheck Flexikon. https://flexikon.doccheck.com/de/Motoneuron_Aufgerufen_am_12.2.23 Aufgerufen am 12.2.23 um 12:44
6) GmbH, D. C. (o. D.-b). Oberes Motoneuron Aufgerufen am 12.2.23. DocCheck Flexikon. https://flexikon.doccheck.com/de/Oberes_Motoneuron_Aufgerufen_am_12.2.23 Aufgerufen am 12.2.23 um 12:55
7) Medizinexpert*innen bei DocCheck. (2022a, Februar 1). Babinski-Reflex - DocCheck. DocCheck Flexikon. https://flexikon.doccheck.com/de/Babinski-Reflex Aufgerufen am 12.2.23 um 13:10
8) A. (2021, 18. April). Klonus: Definition, Ursachen, Tests und Behandlung. Doktor.Top. https://doktor.top/klonus-definition-ursachen-tests-und-behandlung/ Aufgerufen am 12.2.23 um 13:14
9) Biolatto, L. (2022, 19. Dezember). Was ist ein Klonus und was ist die Ursache dafür? Besser Gesund Leben. https://bessergesundleben.de/was-ist-ein-klonus-und-was-ist-die-ursache-dafuer/ Aufgerufen am 12.2.23 um 13:30
10) Medizinexpert*innen bei DocCheck. (2022, 28. Februar). Alpha-Motoneuron - DocCheck. DocCheck Flexikon. https://flexikon.doccheck.com/de/Alpha-Motoneuron Aufgerufen am 17.2.23 um 15:02
11) Clonus: Ursachen, Behandlung und mehr. (o. D.). https://presspassblog.com/de/articles/8229-clonus-causes-treatment-and-more Aufgerufen am 12.2.23 um 13:40
12) Ismail Boyraz 1, Hilmi Uysal 2, Bunyamin Koc 1, Hakan Sarman Clonus: definition, mechanism, treatment 2015 Feb;12(1):19-26.
13) Neurologische Untersuchung - Wissen @ AMBOSS. (o. D.). https://www.amboss.com/de/wissen/Neurologische_Untersuchung Aufgerufen am 17.2.23 um 14:27
14) Kinder- und Jugendpsychiatrie by Helmut Remschmidt (2008-09-05). (o. D.).
Die Pods für Reaktion und Schnelligkeit
Autor: Mike Habdank
Das neue Trainingssystem BlazePod für Sport, Fitness und Therapie verbessert die Reaktionsgeschwindigkeit und kognitiven Fähigkeiten. Je nach Programm leuchten die BlazePods in unterschiedlichen Farben und Abfolgen auf. Durch Berühren des Pods wird das Licht deaktiviert und das nächste Licht wird aktiviert. Dadurch bekommt der Patient ein eindeutiges Feedback die Übung richtig ausgeführt zu haben.
Die Lichtsensoren werden über eine App gesteuert. In der App können vordefinierten Aktivitäten ausgewählt werden oder persönliche Trainingspläne zusammengestellt werden.
Reaktionszeiten und die Anzahl der Berührungen werden in der App aufgezeichnet und stehen zur Analyse zur Verfügung. Die App liefert alle Treffer und Fehlversuche der Pods inklusive Milisekundengenauer Angabe der Reaktionszeit. So lässt sich jeder Trainings erfolg unmittelbar feststellen. Dieses motiviert den Patienten sich zu steigern und weiterzumachen.
Das Beste am BlazePod ist seine große Vielseitigkeit es kann in jede Art von Training integriert werden.
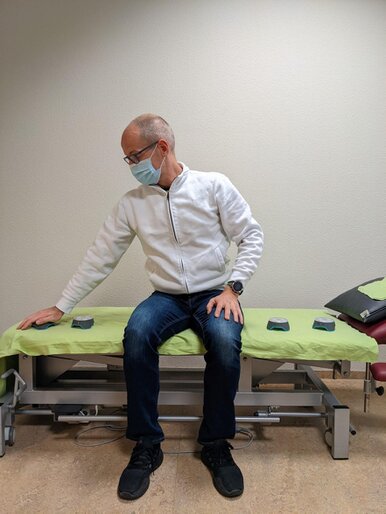
Stützreaktion der Arme im Sitz

Training von Schutzschritten im stehen

Visuelles Cueing und antifreezing Training
bei Morbus Parkinson
Anbahnung der Stifthaltung
Ohne mit einem Stift zu hantieren
Autorin: Janna Materna
Ein häufiges Ziel in der Ergo- und Physiotherapie ist das Führen eines Stiftes zum
Schreiben. Neben Aufgaben mit einem Stift mit/ohne Griffverdickung gibt es
zahlreiche Beispiele für geeignete Übungen.
Es folgen vier Übungsbeispiele
1. Holzdübel aufstellen und auf vorgegebenen Punkten positionieren
2. Farbpunkte mit einem Wattestäbchen setzen
3. Perlen mit einem Pinsel „wegkehren“
4. Stecknadeln stecken
Assessments in der Rehabilitation: Neurologie
Download von verschiedene Assessments auf Deutsch,
in Bereich:
- Frühphase
- Selbstständigkeit im Alltag
- Zielsetzung
- Mobilität und Fortbewegung
- Obere Extremitäten
- Gleichgewicht und Sturzrisiko
- Sensorische Funktionen
- Neurologischer Status und motorische Funktionen
- Kognitive Funktionen und Wahrnehmung
- Krankheitsspezifische Messungen
Apps
Brisa
Brisa ist dein Begleiter im Alltag mit Multipler Sklerose.
Tracke Symptome, Befinden und Aktivitäten, und verstehe, was dir gut tut – so gestaltest du dein Leben mit MS selbstbestimmt.
Cura Swing
App, die das Mitschwingen der Arme beim Gehen in Musik übersetzt und den gesamten Bewegungsablauf stimuliert.
Größere und schnellere Bewegungen steigern automatisiert in Echtzeit die musikalische Intensität. Diese innovative Form des musikalischen „Biofeedbacks“ basiert auf neurowissenschaftlichen Erkenntnissen.
Digitale Hausapotheke
Mithilfe der Digitalen Hausapotheke verwalten Sie Ihren Medikamentenbestand einfach, übersichtlich und effektiv.
Haben Sie immer im Blick welche Einnahmen als nächstes anstehen, was Sie bereits eingenommen haben und welche Medikamente zur Neige gehen, oder bald ablaufen. Erstellen Sie mit wenigen Klicks Ihren persönlichen Medikationsplan zur Vorlage bei Ihrem Arzt oder Apotheker.
Emendia MS
Emendia begleitet Deine Therapie digital. Erhalte über das integrierte Medizinprodukt Emendia MS Therapieunterstützung bei Multipler Sklerose und bleibe gleichzeitig im Austausch mit Deiner Ärztin/ Deinem Arzt.
MoveApp
MoveApp – die Unterstützung ihrer Bewegungstherapie für Parkinson.
Die App bietet ihnen multimediale Informationen über Ursachen und Hintergründe zur Parkinson Erkrankung. Übersichtlich zusammengefasst können sie aus weiteren Bereichen wählen: Übungen, über 20 Videos und Anleitungen zum Bewegungstraining.
My therapy
MyTherapy ist eine Medikamentenerinnerung für die Einnahme Ihrer Tabletten, Pillen oder anderer Medikamente.
Die deutsche Gesundheitsapp unterstützt Sie als Medikamenten Manager und Tagebuch. Die kostenlose Health App hilft Ihnen durch Medikamentenerinnerung per Alarm Ihre Arzneimittel zu nehmen und verwaltet die gesamte Medikamenten-Einnahme, während Sie ganz automatisch RedPoints für vergünstigte Einkäufe bei Shop Apotheke sammeln. Jede Medikamenten-Einnahme lässt sich zur Vorbereitung für das nächste Arztgespräch einfach als Bericht ausdrucken.
Stabyl
App wurde in Zusammenarbeit mit der Deutschen Parkinson-Gesellschaft entwickelt und orientiert sich eng an den Bedürfnissen von Menschen mit der Parkinson-Krankheit. So erkennt die App über den Bewegungssensor im iPad das Handzittern des Nutzers und gleicht es durch virtuelle Gegenbewegungen aus, was die Browseransicht stabilisiert. Wie stark Staybl auf das Zittern reagieren soll, lässt sich individuell einstellen.
StudyMyTremor, Tremor App
Mit StudyMyTremor erfassen Sie das Zittern Ihrer Hände, den Tremor, zuverlässig und korrekt.
Die App setzt eine bisher ausschließlich im professionellen klinischen Umfeld verwendete Methode in eine einfach bedienbare Anwendung um.
Swallow Prompt
Eine personalisierte Erinnerungs-App, die entwickelt wurde, um Personen mit Erkrankungen wie Parkinson, Zerebralparese und anderen gesundheitlichen Herausforderungen zu helfen, die eine übermäßige Speichelproduktion verursachen.
Übernehmen Sie die Kontrolle über Ihre Mundgesundheit und verbessern Sie Ihren täglichen Komfort mit maßgeschneiderten Warnungen und praktischen Vorschlägen.
Voice Trainer
Der Voice Trainer gibt anhand eines Strichs auf dem Telefon eine visuelle Rückmeldung hinsichtlich der Lautstärke und der Tonhöhe wieder. In der Logopädie kann man gemeinsam die erwünschte Lautstärke und Tonhöhe eingestellt werden. Der Voice Trainer kann sowohl zum Trainieren als auch als Kontrolle während eines Gesprächs genutzt werden